
In modern healthcare and medicine, the quest for novel treatments and therapies is a perpetual challenge. Groundbreaking solutions can be discovered not only through innovation but also by uncovering hidden relationships within existing data. This is what Graph Neural Networks (GNNs) do. This cutting-edge fusion of graph theory and deep learning can transform drug repurposing—the process of finding new medical uses for already existing drugs.
Imagine if we could reposition existing drugs to combat diseases they were never originally intended to treat. This is a research question that scientists at Elsevier, a leading provider of technological solutions for medical researchers and healthcare professionals, are trying to address. Drug repurposing can accelerate drug discovery and thus save more lives. GNNs enable us to analyze complex networks of drug-disease interactions and predict new, unexpected connections.
In this blog post, we provide an overview of the key concepts of GNNs and their applications in drug repurposing. We also present a concrete proposal for a GNN implementation aimed at addressing the drug repurposing problem put forward by our colleagues at Elsevier.
What are GNNs?
Graph Neural Networks (GNNs) are a type of neural network designed to operate on data structured as graphs, where they process information from nodes and edges to learn representations and perform tasks specific to the graph’s structure and content.
In drug repurposing, GNNs are used for link prediction.
Link prediction is a task in graph theory and network analysis where the goal is to estimate the likelihood of a link (or connection) existing between two nodes in a graph, given the current structure and features of the graph.
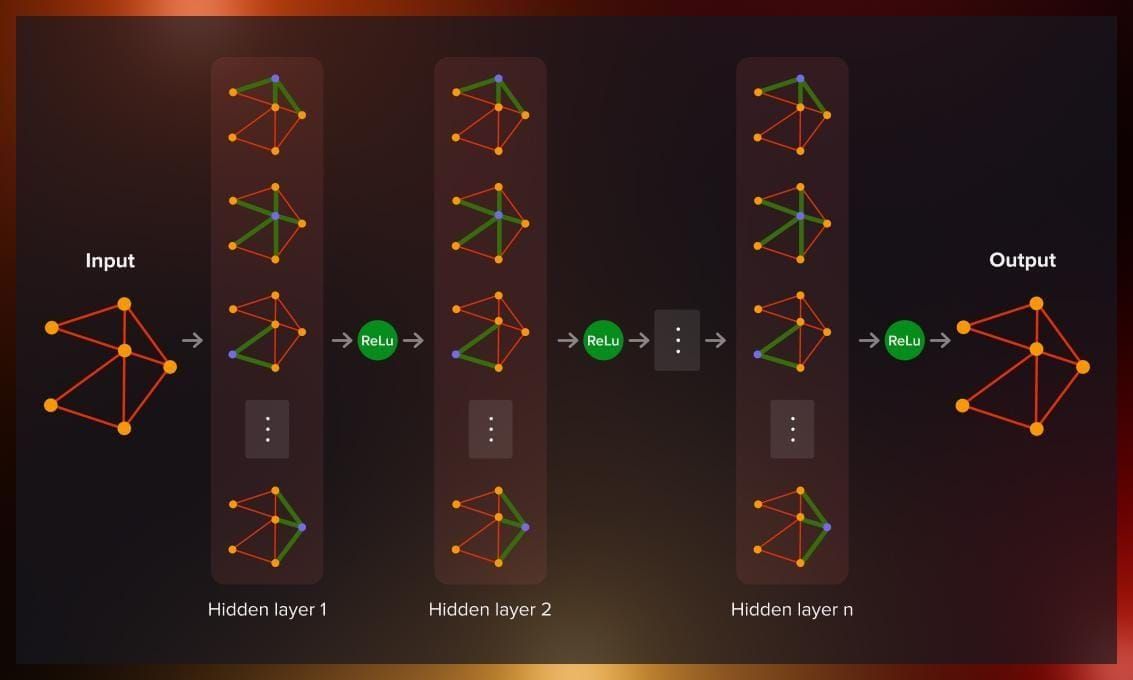
Key concepts of GNNs
Core GNN concepts for link prediction include:
Node representations: Nodes within a graph are associated with feature vectors that evolve through iterative updates. These evolving node representations form the foundation of GNNs’ predictive capabilities.
Message passing: It refers to the process of nodes sending (passing) information (messages) to their neighbors and possibly updating their states based on received messages.
Aggregation and propagation: GNNs employ a message-passing mechanism, enabling the aggregation of information from neighboring nodes. This approach captures both local and global dependencies within the graph, allowing GNNs to model complex relationships.
Graph convolution: Analogous to convolution in CNNs but adapted for graphs. It involves updating node representations by combining their features with those of their neighbors.
What is DTINet?
DTINet (Drug-Target Interaction Network) is a deep learning framework for predicting drug-target interactions. It utilizes the information from diverse types of biological data sources, including known drug-target interactions, drug structures, drug-induced gene expression profiles, protein sequences, and protein-protein interaction networks. By combining these diverse data sources, DTINet can infer potential drug-target interactions more accurately.
DTINet dataset for drug repurposing
Several organizations own private data sets related to a problem they are trying to address. researchers at Elsevier have access to a private dataset that contains relationships, features and interactions of drugs, proteins, diseases, as well as side effects.
Due to the non-disclosure agreement protecting personal data, we do not have access to the Elsevier dataset. However, in this work we rely on the DTINet dataset that possesses similar characteristics.
The DTINet dataset encompasses six crucial networks. Below we explain how they are employed for drug repurposing.
-
Drug-Protein Interaction Network captures the complex interactions between drugs and proteins, forming the foundation for many drug repurposing predictions.
-
Drug-Drug Interaction Network helps how drugs interact with each other, which is essential for identifying potential synergistic effects and drug combinations.
-
Protein-Protein Interaction Network provides critical insights into disease pathways and potential drug targets.
-
Drug-Disease Association Network helps identify new connections between drugs and diseases that can lead to seminal therapeutic discoveries.
-
Protein-Disease Association Network determines proteins associated with specific diseases, which aids in target identification.
-
Drug-Side Effect Network predicts potential side effects of drugs, ensuring patient safety.
-
Disease-Symptoms, Disease-Biological processes, Disease-Clinical Parameters networks help establish similarities between diseases.
-
Protein-Biological Processes, Drug-Symptoms, Drug-Processes help better determine the role of proteins in disease and the effect of the drug on it. They also play a significant role in predicting disease through proteins’ effects on disease-associated parameters.
How do GNNs work for link prediction with GNNs?
The process of link prediction with GNNs involves several key stages:
-
Initialization: Nodes are assigned initial feature vectors, typically based on domain-specific knowledge or randomly.
-
Message passing: Information is iteratively propagated through the graph. Each node receives messages from its neighbors and updates its representation accordingly.
-
Aggregation: Messages are aggregated using functions like weighted sums or attention mechanisms, resulting in new representations for nodes.
-
Update: The aggregated information is used to update the feature vectors of each node.
-
Output: The final node representations can be employed for downstream tasks, such as link prediction.
How are GNNs used in drug repurposing?
Now, let’s look at the process of loading and preprocessing the DTINet dataset, which contains information about drug-disease interactions and drug similarity scores, to create a link prediction GNN that can be applied for drug repurposing.
Step 1. Setup
First, we set up the environment and install the required packages. You can follow along with the complete code in the original notebook.
# Import necessary libraries and check Torch version
import torch
from torch import Tensor
print(torch.__version__)
# Install required packages.
import os
os.environ['TORCH'] = torch.__version__
!pip3 install torch-scatter -f https://data.pyg.org/whl/torch-${TORCH}.html
!pip3 install torch-sparse -f https://data.pyg.org/whl/torch-${TORCH}.html
!pip3 install pyg-lib -f https://data.pyg.org/whl/nightly/torch-${TORCH}.html
!pip3 install git+https://github.com/pyg-team/pytorch_geometric.git
Step 2. Understanding the data
Our dataset comprises a drug-disease interaction network, where nodes represent drugs and diseases, and edges indicate potential interactions between them. we need to organize this data into a format conducive to learning. Here’s a breakdown of our dataset:
-Drug nodes: Each node in the network represents a drug, and we have information about these drugs in the form of features or similarity scores. This similarity score can be generated by embedding drug-target, target-disease, and other relevant networks mentioned above.
-
Disease nodes: Similarly, nodes in the network correspond to diseases, with their respective features.
-
Edges: Edges between drug and disease nodes signify potential interactions. These interactions are valuable for predicting novel drug-disease associations.
Step 3. Data loading
The dataset is typically available in various formats, such as CSV, JSON, or in our case, a custom format stored in a compressed tarball. This is how we load the data:
import tarfile
import numpy as np
import pandas as pd
# Open the compressed tarball containing our data
tar = tarfile.open("./data.tar.gz", "r:gz")
# Load the drug-disease interaction matrix
mat_drug_disease_file = tar.extractfile("data" + "/" + "mat_drug_disease.txt")
mat_drug_disease = np.loadtxt(mat_drug_disease_file)
# Load drug similarity scores
drug_sim_file = tar.extractfile("data" + "/" + "Similarity_Matrix_Drugs.txt")
drug_sim = np.loadtxt(drug_sim_file)
By extracting these matrices, we obtain valuable information about drug-disease interactions and drug similarities.
Step 4. Data transformation
Next, we transform our data to make it suitable for GNN-based link prediction.The process is as follows:
1. Creating DataFrames
We convert the interaction matrix into pandas DataFrames, which simplifies data manipulation and provides a clearer view of the data.
edges_df = pd.DataFrame(data=mat_drug_disease.astype(int),
index=[id_ for id_ in range(np.shape(mat_drug_disease)[0])],
columns=[id_ for id_ in range(np.shape(mat_drug_disease)[1])])
2. Reshaping and filtering
Next, we reshape and filter the DataFrame to create a more structured view of interactions, removing self-links and non-existing links.
edges_df = edges_df.rename_axis('drugId') \
.reset_index() \
.melt('drugId', value_name='link_existence', var_name='diseaseId') \
.query('drugId != diseaseId') \
.reset_index(drop=True).sort_values(by='drugId', ascending=True)
# Filter out non-existing links
edges_df = edges_df.loc[~((edges_df['link_existence'] == 0))]
3. Creating node features
We also create node feature matrices for drugs based on their similarity scores.
drug_features_df = pd.DataFrame(data=drug_sim,
columns=[f"feat_{id_}" for id_ in range(np.shape(drug_sim)[0])])
Step 4. Data mapping
To ensure consistency and efficiency in the GNN model, we map unique drug and disease IDs to consecutive values, thus simplifying indexing and processing.
`python
# Mapping of drug IDs to consecutive values
unique_drug_id = edges_df['drugId'].unique()
unique_drug_id = pd.DataFrame(data={
'drugId': unique_drug_id,
'mappedID': pd.RangeIndex(len(unique_drug_id)),
})
# Mapping of disease IDs to consecutive values
unique_disease_id = edges_df['diseaseId'].unique()
unique_disease_id = pd.DataFrame(data={
'diseaseId': unique_disease_id,
'mappedID': pd.RangeIndex(len(unique_disease_id)),
})
With our data loaded, transformed, and mapped, we’re now ready to construct the graph structure and start building and training our GNN model for drug repurposing. In the upcoming sections, we’ll look at constructing the graph and implementing the GNN architecture to predict potential drug-disease interactions.
Step. 5 Edge index construction
The backbone of any graph-based analysis is the construction of the edge index, which defines how nodes are connected within the graph.
The edge index, often referred to as the “adjacency matrix” in traditional graph theory, outlines the relationships between nodes. For our task, we’re particularly interested in establishing connections between drugs and diseases. These connections signify potential interactions, forming the foundation for the GNN’s learning process.
# Perform merge to obtain the edges from drugs and diseases
interactions_drug_id = pd.merge(edges_df['drugId'], unique_drug_id,
left_on='drugId', right_on='drugId', how='left')
interactions_drug_id = torch.from_numpy(interactions_drug_id['mappedID'].values)
interactions_disease_id = pd.merge(edges_df['diseaseId'], unique_disease_id,
left_on='diseaseId', right_on='diseaseId', how='left')
interactions_disease_id = torch.from_numpy(interactions_disease_id['mappedID'].values)
# Construct edge index for drug to disease interactions
edge_index_drug_to_disease = torch.stack([interactions_drug_id, interactions_disease_id], dim=0)
assert edge_index_drug_to_disease.size() == (2, 199079)
# Display the final edge indices pointing from drugs to diseases
print("Final edge indices pointing from drugs to diseases:")
print("=================================================")
print(edge_index_drug_to_disease)
With our data prepared, it’s time to build and train a Graph Neural Network (GNN) for drug repurposing.
GNN architecture
Now let’s look at the GNN architecture. Designed with simplicity and effectiveness in mind, it has just a few key layers. Here’s an overview of its architecture:
1. Node embeddings
At the heart of the GNN are node embeddings. In drug repurposing, these embeddings represent drugs and diseases in a lower-dimensional space, making them amenable to processing by the neural network.
For drugs, we utilize two components:
-
A linear layer (
self.drug\_lin) that learns from existing features. -
An embedding layer (
self.drug\_emb) for further representation learning.
Similarly, we have embeddings for diseases, and these embeddings serve as the foundation of the GNN’s understanding of the graph.
2. Heterogeneous Graph Convolutional Network
To analyze and propagate information through the heterogeneous graph, we employ a custom Graph Convolutional Network (GCN) architecture. Specifically, we use the SAGEConv (GraphSAGE) layers, which are designed for inductive, scalable graph learning.
Our GNN comprises two SAGEConv layers (self.conv1 and self.conv1) stacked in sequence. These layers allow the model to capture complex relationships between drugs and diseases by aggregating information from neighboring nodes.
from torch_geometric.nn import SAGEConv, to_hetero
class GNN(torch.nn.Module):
def __init__(self, hidden_channels):
super().__init__()
self.conv1 = SAGEConv(hidden_channels, hidden_channels)
self.conv2 = SAGEConv(hidden_channels, hidden_channels)
def forward(self, x: Tensor, edge_index: Tensor) -> Tensor:
x = F.relu(self.conv1(x, edge_index))
x = self.conv2(x, edge_index)
return x
3. Classifier
We also need a classifier to make predictions based on the GNN’s output. Our classifier takes drug and disease embeddings and calculates edge-level predictions.
class Classifier(torch.nn.Module):
def forward(self, x_drug: Tensor, x_disease: Tensor, edge_label_index: Tensor) -> Tensor:
edge_feat_drug = x_drug[edge_label_index[0]]
edge_feat_disease = x_disease[edge_label_index[1]]
return (edge_feat_drug * edge_feat_disease).sum(dim=-1)
Model integration
To build the final model, we integrate the GNN and classifier components while also considering embeddings for drugs and diseases.
class Model(torch.nn.Module):
def __init__(self, hidden_channels):
super().__init__()
self.drug_lin = torch.nn.Linear(709, hidden_channels)
self.drug_emb = torch.nn.Embedding(data["drug"].num_nodes, hidden_channels)
self.disease_emb = torch.nn.Embedding(data["disease"].num_nodes, hidden_channels)
self.gnn = GNN(hidden_channels)
self.gnn = to_hetero(self.gnn, metadata=data.metadata())
self.classifier = Classifier()
def forward(self, data: HeteroData) -> Tensor:
x_dict = {
"drug": self.drug_lin(data["drug"].x) + self.drug_emb(data["drug"].node_id),
"disease": self.disease_emb(data["disease"].node_id),
}
x_dict = self.gnn(x_dict, data.edge_index_dict)
pred = self.classifier(
x_dict["drug"],
x_dict["disease"],
data["drug", "interactions", "disease"].edge_label_index,
)
return pred
# Initialize the model
model = Model(hidden_channels=64)
Training the model
To train our model, we use binary cross-entropy loss and the Adam optimizer. The training loop iterates over the dataset for a specified number of epochs.
import tqdm
import torch.nn.functional as F
# Set the device (GPU if available, otherwise CPU)
device = torch.device('cuda' if torch.cuda.is_available() else 'cpu')
model = model.to(device)
optimizer = torch.optim.Adam(model.parameters(), lr=0.001)
# Training loop
for epoch in range(1, 6):
total_loss = total_examples = 0
for sampled_data in tqdm.tqdm(train_loader):
optimizer.zero_grad()
sampled_data.to(device)
pred = model(sampled_data)
ground_truth = sampled_data["drug", "interactions", "disease"].edge_label
loss = F.binary_cross_entropy_with_logits(pred, ground_truth)
loss.backward()
optimizer.step()
total_loss += float(loss) * pred.numel()
total_examples += pred.numel()
print(f"Epoch: {epoch:03d}, Loss: {total_loss / total_examples:.4f}")
Model validation and evaluation
Now that our model is trained, we need to validate its performance and evaluate its ability to predict drug-disease interactions. For that, we split our dataset into training, validation, and test sets. This is how we work with the validation set:
# Define the validation seed edges
edge_label_index = val_data["drug", "interactions", "disease"].edge_label_index
edge_label = val_data["drug", "interactions", "disease"].edge_label
val_loader = LinkNeighborLoader(
data=val_data,
num_neighbors=[20, 10],
edge_label_index=(("drug", "interactions", "disease"), edge_label_index),
edge_label=edge_label,
batch_size=3 * 128,
shuffle=False,
)
One common metric used for binary classification tasks is the Area Under the Receiver Operating Characteristic Curve (AUC-ROC). AUC-ROC measures the model’s ability to distinguish between positive and negative examples. An AUC value closer to 1.0 indicates excellent performance, while an AUC value of 0.5 suggests random guessing.
We achieved the validation AUC of 0.9. This indicates that our GNN-based model performs exceptionally well in distinguishing potential drug-disease interactions. Our AUC implies that the model has a high true positive rate while maintaining a low false positive rate, which is precisely what we want in drug repurposing tasks.The high AUC score demonstrates the model’s capability to predict new interactions between drugs and diseases effectively.
In addition to AUC-ROC, other evaluation metrics, such as precision, recall, and F1-score, can provide a more comprehensive understanding of the model’s performance, especially if there are imbalances in the dataset. These metrics can help you fine-tune the model or make decisions about its practical applications.
from sklearn.metrics import roc_auc_score
preds = []
ground_truths = []
for sampled_data in tqdm.tqdm(val_loader):
with torch.no_grad():
sampled_data.to(device)
preds.append(model(sampled_data))
ground_truths.append(sampled_data["drug", "interactions", "disease"].edge_label)
pred = torch.cat(preds, dim=0).cpu().numpy()
ground_truth = torch.cat(ground_truths, dim=0).cpu().numpy()
auc = roc_auc_score(ground_truth, pred)
print(f"Validation AUC: {auc:.4f}")
Conclusion
Graph Neural Networks have a significant potential in drug discovery, specifically in drug repurposing. In this blog post, we have demonstrated the use of GNNs, using Serokell’s work as an example. Our model, evaluated on a test dataset, demonstrated a validation AUC of 0.9. This result signifies the model’s capacity to predict potential drug-disease interactions with a high degree of accuracy.
By operating on graph-structured data, GNNs can analyze molecular structures and intricate biological networks. This capability allows for the detailed examination of relationships within biomolecular graphs, helping find potential new uses for existing drugs. With GNNs, the process of drug repurposing becomes data-driven, which significantly reduces time for drug discovery and opens up wide opportunities for science.



.jpg)
.jpg)

